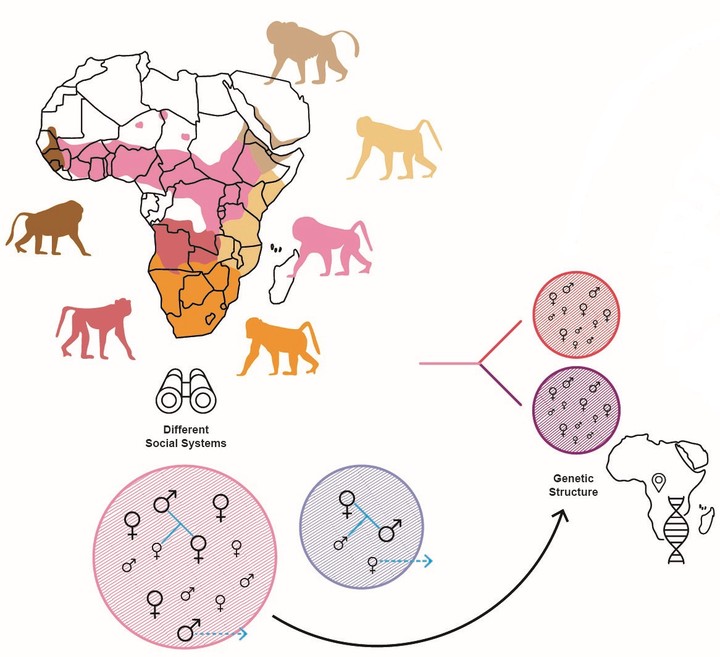
There is ample evidence that a species’ social system influences its population genetic structure, for example through effects on gene flow (e.g. dispersal behaviour) or effective population size and genetic drift (e.g. reproductive skew). However, empirical population genetic studies are usually site- and species-specific. Population genetic studies that investigate behavioural traits commonly concentrate on local populations and are more limited in range than studies that investigate the impact of ecological or landscape parameters on genetic structure and diversity, thereby neglecting broader macroevolutionary impacts. We are lacking a general understanding about how consistentlybehavioural traits influence distribution-wide genetic structure and diversity across taxa. This link is pivotal if we want to scrutinize the role of behavioural traits in macroevolutionary processes. I will examine this link along two lines, taking advantage of the accumulation of DNA sequence information as well as recent developments in population genomics of wild populations of non-model organisms. First, I will geo-reference DNA sequences from open databases (GenBank and GBIF) across a diverse set of animal taxa to derive standardized estimates of distribution-wide genetic diversity and structure and combine these with quantitative measures of social systems. I will employ novel machine learning approaches to unravel important predictors of genetic structure and diversity, taking into account various potential confounding variables. Secondly, I will conduct population genomic studies in some wild animal populations to get a comprehensive view of the distribution of genetic variation on the genome level over the scales from the group to the local population. A number of population genomic methods have been developed very recently to specifically estimate relatedness from very low-coverage sequencing data. While they mainly targeted studies of ancient and prehistoric human remains, they also hold great promise for studies of natural animal populations, especially if samples can only be obtained non- invasively and the resulting DNA holds the same pitfalls as ancient DNA (aDNA), i.e. very low DNA quality and quantity as well as extreme levels of contamination. The advantages of this novel genome- scale approach over classical markers (e.g. microsatellites) are the better comparability across studies (no ascertainment bias), the higher accuracy in relatedness estimation, and the increased power to detect shallow differences. I will employ low-coverage shot-gun sequencing or, in cases where only non-invasive samples can be obtained, whole-genome hybridization capture and estimate dyadic relatedness among individuals using algorithms that account for sequence uncertainty. Detailed behavioural data on movement and individual interactions to address the relationship between behavioural measures and genetic structure will be contributed by my collaboration partners.
Addressing the behavioural drivers of population genetic structure